
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Coordinate Statement Previous: Coordinate Statement
Syntax
The coordinate statement options are carried out when the END statement is issued. If one wants to carry out several coordinate manipulations, each manipulation has to be initiated separately from the main level of X-PLOR.- COORdinates
 coordinate-statement
coordinate-statement END
END - is invoked from the main level of X-PLOR. The END statement activates execution of the particular operation.
- coordinate-statement:==
-
- COPY
- [SELEction=selection] copies main coordinate
set into comparison set;
XCOMP
 X,YCOMPY,ZCOMPZ; B,Q are
unaffected (default for selection: (ALL) ).
X,YCOMPY,ZCOMPZ; B,Q are
unaffected (default for selection: (ALL) ).
- FIT
- { [SELEction=selection]
[MASS=logical]
[LSQ=logical] }
rotates (if LSQ is TRUE) and translates all main coordinates
to obtain a best fit between the
selected main and
comparison atoms.
Translation superimposes
the geometric centers (or the centers of mass if MASS is TRUE)
of the two coordinate sets. Rotation
occurs with the Kabsch (1976)
least-squares fitting algorithm. Mass-weighting
is applied if MASS is set to TRUE. Upon successful
completion of this operation, the Eulerian
angles describing the rotation matrix 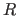 are stored in the symbols
$THETA1, $THETA2, $THETA3, and the translation vector
are stored in the symbols
$THETA1, $THETA2, $THETA3, and the translation vector
 is stored
in the symbols $X, $Y, $Z.
The fitted coordinate set
is stored
in the symbols $X, $Y, $Z.
The fitted coordinate set  is
related to original set
is
related to original set 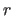 by
by  (default for
selection: (ALL); for MASS: FALSE; for LSQ: TRUE).
(default for
selection: (ALL); for MASS: FALSE; for LSQ: TRUE).
- FRACtionalize
- { [SELEction=selection]
[A=real] [B=real] [C=real]
[ALPHa=real]
[BETA=real] [GAMMa=real] }
fractionalizes selected coordinates. The X-PLOR
convention
keeps the direction of the x-axis
(x same direction as a; y is in (a,b) plane)
and is the same that is used
internally by all X-PLOR routines
(default for selected atoms: (ALL);
for a, b, c: 1.0; for
 : 90
: 90 ).
).
- INITialize
- { [SELEction=selection] }
initializes main coordinate
set; i.e., X:=9999.0, Y:=9999.0, Z:=9999.0; B,Q are
unaffected (default for selection:
(ALL) ).
- ORIEnt
- { [SELEction=selection]
[MASS=logical]
[LSQ=logical] }
rotates (if LSQ is TRUE) and translates all
coordinates such that
the principal axis system of the selected atoms
corresponds to the x,y,z axis. The translation superimposes
the geometric center (or the center of mass if MASS is TRUE)
and the coordinate origin. The rotation
occurs with the Kabsch (1976)
least-squares fitting algorithm. Mass-weighting
is applied if MASS is set to TRUE.
Upon successful
completion of this operation, the Eulerian
angles describing the rotation matrix are stored in the symbols
$THETA1, $THETA2, $THETA3, and the translation vector
is stored
in the symbols $X, $Y, $Z.
The oriented coordinate set is
related to original set by
(default for
selection: (ALL); for MASS: FALSE; for LSQ: TRUE).
- ORTHogonalize
- { [SELEction=selection]
[A=real] [B=real] [C=real]
[ALPHa=real] [BETA=real] [GAMMa=real] }
orthogonalizes selected coordinates. The X-PLOR
convention
keeps the direction of the x-axis
(x same direction as a; y is in (a,b) plane) and is the same that is used
internally by all X-PLOR routines
(default for selected atoms: (ALL);
for a, b, c: 1.0; for
: 90).
- RGYRation
- { [SELEction=selection]
[MASS=logical]
[FACT=real] }
computes radius of gyration
where the angle brackets denote averaging over selected atoms. The averaging is mass-weighted if MASS is TRUE. The factor FACT is subtracted from the masses before applying the mass-weighting. The symbols $RG (radius of gyration), $XCM, $YCM, $ZCM (center of mass) are declared (default for selected atoms: (ALL); for MASS: FALSE; for FACT: 0.0).
(6.1) - RMS
- { [SELEction selection]
[MASS=logical] }
computes the (mass-weighted if MASS is TRUE)
rms difference for
selected atoms between the main and comparison set. The
rms value is stored in the symbol $RESULT. The
individual atomic rms differences are stored in
the RMSD array
(default for selection: (ALL); for MASS: FALSE).
- ROTAte
- { [SELEction=selection]
[CENTer=3d-vector] matrix }
rotates selected atoms around the specified
rotation center (default: ( 0 0 0) ).
The rotation matrix is specified through the
matrix statement (see Section 2.4)
(default for
selection: (ALL)).
- SHAKe
- { [MASS=logical] [REFErence=MAIN
 COMP] }
iteratively modifies main coordinate set until SHAKE
constraints (see Section 8.2) are satisfied. The reference
coordinate set specifies the direction of the SHAKE shift
vectors
(default for MASS: FALSE; for REFErence: MAIN).
COMP] }
iteratively modifies main coordinate set until SHAKE
constraints (see Section 8.2) are satisfied. The reference
coordinate set specifies the direction of the SHAKE shift
vectors
(default for MASS: FALSE; for REFErence: MAIN).
- SWAP
- { [SELEction=selection] } exchanges main
and comparison
coordinate set, i.e.,
X,Y,Z
 XCOMP,YCOMP,ZCOMP; B,Q are unaffected
(default for selection: (ALL) ).
XCOMP,YCOMP,ZCOMP; B,Q are unaffected
(default for selection: (ALL) ).
- SYMMetry
- symmetry-operator
[SELEction=selection]
applies the specified
crystallographic symmetry operator
to the selected coordinates.
The notation for the symmetry operator
is the same as in the International Tables for
Crystallography (Hahn, 1987),
e.g., (
 ,
,  ,
,  ). The unit cell
geometry needs to be specified (Section
13.3) before invoking this statement. The coordinates
are converted into fractional coordinates before application
of the symmetry operator and converted back into orthogonal
coordinates afterward.
). The unit cell
geometry needs to be specified (Section
13.3) before invoking this statement. The coordinates
are converted into fractional coordinates before application
of the symmetry operator and converted back into orthogonal
coordinates afterward.
- TRANslate
- { [SELEction=selection]
VECTor=3d-vector [DISTance=real] }
translates selected atoms by specified translation
vector. If DISTance is specified, the translation occurs along
the specified vector for the specified distance (default for
selection: (ALL) ).
- COOR coordinate-read-statement END
- reads coordinates. It is invoked from the main level of X-PLOR.
- coordinate-read-statement:==
- [DISPosition= COMParison
DERIvative MAIN
REFErence ]
[SELEction=selection]
{pdb-record }
reads Brookhaven Data Bank formatted records consisting
of x,y,z coordinates, occupancies, and B-factors,
tries to match the atom name, residue name,
residue number, and
segment name, and
deposits the information in the main (X,Y,Z,B,Q), comparison
(XCOMP, YCOMP, ZCOMP, BCOMP, QCOMP),
reference (REFX, REFY, REFZ, HARM, HARM),
or derivative (DX, DY, DZ, FBETA, FBETA) coordinate arrays. (For the
definition of these arrays, see Section 2.16.) The
information is deposited only if the atom has been selected.
Note that the syntax is strict; i.e., the SELEction
and the DISPosition
have to be specified before one can read a pdb-record. Also,
the PDB convention suggests an END statement at the end of the file
that will terminate the coordinate statement
(default for selected atoms: (ALL); for DISPosition: MAIN).
- pdb-record:==
- According to the Brookhaven Protein Data Bank, the entry for atoms is defined as follows:
ATOM 837 HG23 THR 1055 -8.573 5.657 -3.818 1.00 0.00
ATOM 1223 O GLY 153A -11.704 -9.200 .489 1.00 0.80
uuuuu vvvv uuuuCuuuuI vvvvvvvvuuuuuuuuvvvvvvvvuuuuuuvvvvvv iii
atom residue x y z q b entry#
number name name number
^ insertion character
^ chain identifier
^ additional character for some atom names (mostly h's)
X-PLOR does not use the chain identifier information. Instead,
it uses the characters in columns 73-76 for the segment name
(see Section 3.7). The segment name
has to match
the definition in the segment statement. The
insertion character is treated
as part of the residue
number (note: the residue number is a string consisting of
a maximum of four characters).
X-PLOR ignores any
reference to the atom numbers and instead generates its own numbering
scheme. The REMARK record of PDB files is treated as a title
record (cf. REMARKS, Section 2.8). No other type of
PDB specification, such as HETAT, SCALE, or SEQU, is
interpreted at present.
These additional records have to be removed before one reads PDB coordinates
with X-PLOR. Initially, the
user should divide the original PDB file into files
containing individual protein chains, individual substrates,
all waters combined, and individual cofactors. This
will simplify the molecular structure generation with X-PLOR (see Section
3.7). Normally, X-PLOR expects
orthogonal coordinates. The ORTHogonalize option
can be used to convert fractional coordinates into
orthogonal Å coordinates (see Section
13.2).
The PDB convention requires an END statement at the end of the coordinate file. X-PLOR uses the same convention. The inclusion of the END statement implies that the coordinate statement must not be terminated with an END statement from the main level of X-PLOR. However, if the END statement is missing in the coordinate file, parsing errors will result.
Xplor-NIH 2026-07-10