
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Setup of the Relaxation Previous: Setup of the Relaxation
Syntax
- RELAxation {
 relaxation-statement
relaxation-statement } END
} END - is
invoked from the
main level of X-PLOR. - relaxation-statement:==
- ASSIgn
- selection selection {
![${<\mbox{real}>[<\mbox{real}>[<\mbox{real}>]]}$](img790.png) }
adds a single cross-peak intensity or a
buildup curve between the two
specified selected sets of spins. The number of mixing times
read depends on the number of CLASs statements issued before the
ASSIgn statement.
Error estimates can be specified. The
format depends on the ERROr_input statement. For
examples, see Section 39.7.
}
adds a single cross-peak intensity or a
buildup curve between the two
specified selected sets of spins. The number of mixing times
read depends on the number of CLASs statements issued before the
ASSIgn statement.
Error estimates can be specified. The
format depends on the ERROr_input statement. For
examples, see Section 39.7.
- AVERage real
- specifies an exponent for calculating the average distance to an UNREsolved group of protons (default: 6).
- CALIbrate { calibrate-statement } END
- calculates the calibration factor between the observed and calculated intensities. Prior to the calibration, this command computes the NOE intensities from the current atomic model unless VALUe is specified.
- calibrate-statement:==
-
- AUTOmatic=ON,OFF
- sets the calibration factor, if ON, every step during the refinement. At present, this allows the calculation of the gradient only approximately. Therefore, it should be turned OFF during minimization.
- GROUp=CLASs,GROUp,ALL
- for CLASS, calculates the calibration factor separately for each classification; for ALL, one overall calibration factor is used (default: CLASs).
- QUALity=real
- specifies that cross peaks for which the relative error estimate exceeds the specified value are not used (default: 1).
- REFErence=ALL,REFErence
- for ALL, uses all peaks that meet the criterion set with the EXCLude option for the calculation of the calibration factor; for REFErence, uses only those that were flagged as reference peaks during input (default: ALL).
- VALUe class *real*
- sets a value for the calibration factor. Note: if the calibration factor is set by VALUe for a class, it is not recalculated for other classes in the same CALIbrate statement.
- AUTOmatic=
- CLASsification class-name
- partitions an intensity table; it applies to all following ASSIgn entries until another CLASS entry is issued. In contrast to the NOE CLASs statement, the RELAxation CLASs statement can be used to switch between classes during the input of cross-peak intensities. Several class statements can be issued without an intervening ASSIgn statement. This allows the input of buildup curves (default: NIL).
- CLGR (CLass_GRoup) *class* group
- enters a CLASs into a GROUp of spectra (default: none).
- CUTOff
- { cutoff-statement } END applies a cutoff to
relaxation matrix and/or gradient calculation. When a cutoff is in
effect, for each peak
on the data list a separate small relaxation matrix is set up,
including only spins
within a range of the spin pair specified by the cutoff.
The spectrum and the gradient are calculated with this
small relaxation matrix.
- cutoff-statement:==
-
- MODE=NONE,GRADient,ALL
- applies no cutoff, cutoff only for the gradient calculation, or cutoff for both intensity and gradient calculation (default: NONE).
- RELAtive_value=real
- specifies a cutoff relative to the actual distance between the two spins in Å (default: 0).
- VALUe=real
- specifies the actual value of the cutoff, in Å (default: 6).
- MODE=
- EEXPonent real
- is an exponent
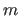 of the WELL or
PARAbolic energy function (default: 2).
of the WELL or
PARAbolic energy function (default: 2).
- ERROr_input
- { error-input-statement } END
defines an error input format for the following ASSIgn statement.
It remains in effect until another ERROr_input command is
issued.
- errorinput-statement:==
-
- INPUt=UNIForm,RANGe,PLUSminus
- specifies the input format (0, 1, or 2 error estimates) for the following ASSIgn statements (default: RANGe): for UNIForm, no error estimate is expected by the program, for RANGe, one error estimate, and for PLUSminus, two, where the first is an error estimate on the minus side, the second on the plus side.
- MODE=ABSOlute,RELAtive
- interprets the error estimate in the ASSIgn statements as absolute or relative (default: ABSOlute).
- VALUe=real
- enters the actual value for UNIForm option (default: 0).
- INPUt=
- GROUp group-name
- defines a group of several
CLASses that are recorded with the same physical conditions,
like solvent(H
 O, DO), field strength, and temperature
(default: NIL).
O, DO), field strength, and temperature
(default: NIL).
- IEXPonent real
- specifies an exponent
 of the calculated and
observed intensities in the energy function (default: 1).
of the calculated and
observed intensities in the energy function (default: 1).
- IWEIght
- { iweight-statement } END applies
individual weights
 to each single term in the energy function.
At the moment, the only supported options are weights equal to the inverse
of
powers of the observed intensity (i.e.,
to each single term in the energy function.
At the moment, the only supported options are weights equal to the inverse
of
powers of the observed intensity (i.e.,
 ).
If the exponent
).
If the exponent  matches the exponent of the energy
function, the weighting is equivalent to refining relative intensity
differences. The minimum weight applied is always 1.
matches the exponent of the energy
function, the weighting is equivalent to refining relative intensity
differences. The minimum weight applied is always 1.
- iweight-statement:==
-
- CUTOff=real
- is the maximum weight applied (default: 10000).
- EXPOnent=real
- specifies an exponent of the
observed intensity in the weight function
(default: 2).
- QUALity=real
- stipulates that cross peaks for which the relative error estimate exceeds the specified value are not used in determining the maximum weight (default: 1).
- CUTOff=
- MINIntensity *class* real
- sets the minimum detected intensity for each class. The specified value is used as the “zero" for the intensities (default: smallest number used by X-PLOR).
- NREStraints integer
- is the required parameter that specifies the maximum expected number of intensities. It has to be greater than or equal to the actual number. The parameter is used to assign space dynamically for the intensity list (default: 200).
- OCCUpancy group selection real
- sets the occupancy for selected spins (for example, 0.9 for exchanging amid protons) (default: 1.0).
- OMEGa *group* real
- specifies the spectrometer frequency, in Hz
(default: 500*10
 ).
).
- OMIT group selection
- is an obsolete statement that was used in a prerelease version of the relaxation matrix code of X-PLOR. It will be removed in a future version. The statement deletes spins from the relaxation matrix. This is almost the reverse of the SELEct command, but it leaves any existing base of unresolved groups intact (default: none).
- POTEntial WELL,PARAbolic
- provides two alternatives: WELL uses a flat-bottom potential with bounds at error estimates, PARAbolic a (generalized) parabola with exponent EEXP and center at the specified intensity (default: WELL).
- PREDict_intensities
- class
{ predict-intensities-statement } END
calculates a spectrum from coordinates with the parameters valid for the
specified class. Note that this destroys any existing experimental intensity database.
The spectrum can be calculated for parts of a molecular structure.
- PREDict_intensities-statement:==
-
- CUTOFf real
- specifies a distance cutoff for cross-peak calculation (default: 10).
- CUTON real
- specifies a distance cuton for cross-peak calculation (default: 0).
- DIAGonal ON,OFF
- prints diagonal peaks (default: OFF).
- FORMat NORMal,LIST
- is an output format; LIST produces an intensity INPUT file (default: NORMal).
- FROM selection
- selects the first part of a molecular structure (default: all NMR-active atoms).
- THREshold real
- specifies the minimum intensity that is printed (default: 0).
- TO selection
- selects the second part of a molecular structure (default: all NMR-active atoms).
- CUTOFf
- PRINt_deviations
- { print-deviation-statement } END
prints the deviations from observed intensities and calculates
an
 value. It uses the form of the energy function
(WELL or PARAbolic and the value of IEXP) specified
before issuing the statement.
value. It uses the form of the energy function
(WELL or PARAbolic and the value of IEXP) specified
before issuing the statement.
- print-deviation-statement:==
-
- FORMat NORMal,LIST
- is an output format; LIST produces a listing in the format of an intensity input file.
- SELEct selection
- calculates the value with only a
subset of the atoms. This allows the calculation of
local values.
- THREshold real
- specifies the minimum deviation that is printed, in the arbitrary units of the input intensities. For a complete list of deviations (including 0), THREshold should be set to a negative number (default: 0).
- FORMat
- REFErence
- flags intensities read by the following ASSIgn statements as reference peaks for the calculation of the calibration factor (see CALIbrate statement), and remains in effect until another CLASs statement is issued. This flag allows one to use a subset of peaks for the calibration, e.g., peaks that do not depend very much on the conformation of the molecule. The reference peaks are written into a separate file, and the reference flag is set before reading that file.
- RESEt
- erases the current relaxation database.
- SELEct group selection
- selects the “NMR-active" atoms (default: (HYDRogen) ). At present, only protons can be used in the refinement. The NMR parameters for protons are applied to all selected atoms. This command initializes the array that stores information about unresolved groups (see below). If the command is not issued at the beginning of the relaxation matrix setup, all hydrogens are used by default. Since a hydrogen is defined by its mass in X-PLOR, care should be taken in this case to modify the masses of hydrogens for simulated annealing only after the relaxation matrix parameters have been set up.
- SORT_intensities
- sorts the intensity list so that peaks with different mixing times belonging to the same spin pair are consecutive on the list. If a cutoff is used, a new relaxation matrix generally has to be set up and diagonalized for each peak on the data list. However, if the only difference between two entries on the data list is the mixing time, the relaxation matrix does not have to be recalculated and diagonalized. Sorting can thus save a considerable amount of CPU time.
- TAUCorrelation
- group { TAUC-statement } END
specifies the correlation time and the order parameter, depending
on the chosen model.
- TAUCorrelation-statement:==
-
- MODEl RIGId
 LIPAri
LIPAri - provides two alternatives: RIGId expects no order parameter; LIPAri expects one overall correlation time and one order parameter. This applies to both the ISOTropic and the VECTor case.
- ISOTropic
![$<\mbox{real}>[<\mbox{real}>]$](img795.png)
- specifies an isotropic correlation time and an order parameter (default: 10 ns, 1).
- RESEt
- initializes a table.
- VECTor selection selection
- specifies a
correlation time (in seconds) and an order parameter for a specified vector.
- MODEl
- TAUMix *class* real
- specifies the mixing time
 , in seconds (default: 0.1 sec).
, in seconds (default: 0.1 sec).
- TOLErance real
- if not equal to zero,
approximates the exact calculation of the
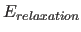 energy function and its first
derivatives with respect to the atomic coordinates
by freezing
until atoms
have moved by less than TOLErance Å from the point at which
was
last computed. This is particularly useful for
molecular dynamics calculations. It should be set to 0
during minimization to avoid inconsistencies
in the force field (default: 0).
energy function and its first
derivatives with respect to the atomic coordinates
by freezing
until atoms
have moved by less than TOLErance Å from the point at which
was
last computed. This is particularly useful for
molecular dynamics calculations. It should be set to 0
during minimization to avoid inconsistencies
in the force field (default: 0).
- UPDate
- computes the NOE intensities from the current atomic model.
- UNREsolved
- group { unresolved-statement } END
replaces protons with unresolved chemical shifts by a single
spin. The distance to an unresolved group is calculated
as
 , where the exponent
, where the exponent  is set with the
AVERage command. A diagonal leakage rate is added
to the relaxation matrix for each unresolved group.
The main use of this command is for methyl groups.
is set with the
AVERage command. A diagonal leakage rate is added
to the relaxation matrix for each unresolved group.
The main use of this command is for methyl groups.
- unresolved-statement:==
-
- 1-2
- treats each methylene group as an unresolved group of protons. A methylene group is recognized by X-PLOR as two hydrogens bound to a heavy atom.
- METHyl
- treats each methyl group as an unresolved group of protons. A methyl group is recognized by X-PLOR as three hydrogens bound to a heavy atom.
- NONE
- deletes all previously defined unresolved groups.
- SELECT selection
- treats the selected atoms as an unresolved group of protons.
- WEIGht *class* real
- specifies the
energy constant
 (overall weight)
(default: 1).
(overall weight)
(default: 1).
- ZLEAkage_rate *group*
- specifies the overall diagonal
leakage rate
 , in sec
, in sec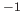 (default: 0).
(default: 0).